
Alkaptonuria (AKU) Unmasked in a Patient with Long-Standing Spondyloarthropathy
Background: Alkaptonuria (AKU) is a rare autosomal recessive metabolic disorder caused by deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), resulting in accumulation of homogentisic acid (HGA). This biochemical defect leads to homogentisic aciduria, ochronosis, and ochronotic osteoarthropathy, with systemic complications that may include valvular heart disease and urolithiasis.
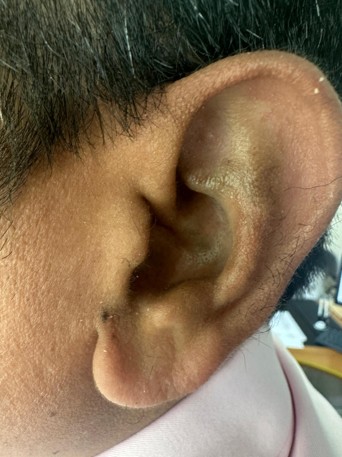
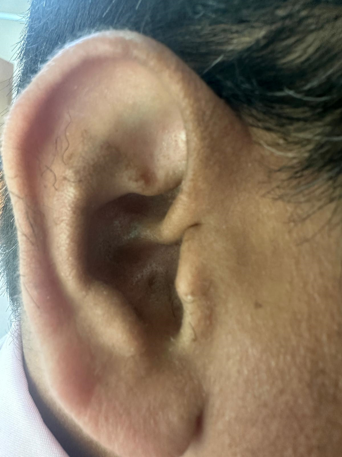
Case Presentation: We report the case of a 37-year-old male with a longstanding history of seronegative spondyloarthropathy and severe osteoarthritis, ultimately requiring bilateral total knee arthroplasty. Despite years of treatment, the underlying etiology was later identified as Alkaptonuria. Key historical clues included dark-colored urine since adolescence and subtle bluish-grey discoloration of the auricular cartilage. Laboratory assessment revealed urine that progressively darkened on standing, consistent with homogentisic aciduria.
Diagnosis and Management: The diagnosis was confirmed through 24-hour urinary HGA quantification using gas chromatography–mass spectrometry (GC–MS). Management focused on supportive therapy, including analgesia, physiotherapy, and joint replacement, along with consideration of Nitisinone—an emerging disease-modifying agent that can reduce HGA production by more than 95%.
Conclusion: This case highlights the importance of recognizing the hallmark triad of AKU: urine darkening, ochronosis, and early-onset osteoarthropathy. Simple historical findings such as chronic dark urine can significantly hasten diagnosis, enabling earlier therapeutic intervention and appropriate genetic counseling for affected families.
Credit: Case managed and reported by Dr. Teerath Kumar, Bahrain Specialist Hospital.


Speak with our Contact Center for assistance